Research
Our research interest is statistical modelling of genomics data and interpretation of genetics variants. Advances in genomics technology have generated, and continue to generate, big data by measuring DNA, RNA, protein, function alongside clinical features including measures of disease activity, progression and related metadata. How to use the advanced artificial intelligence and other computational method to analyze these big data to facilitate precision medicine is currently of great interest. A person’s genome typically contains millions of variants which represent the differences between this personal genome and the reference human genome. It is challenging to understand the mechanism of how these genetic variants contribute to disease or other trait because over 90% of trait-associated genetic variants are located in non-coding regions which don’t encode any protein-coding genes but may invlved in gene regulatory networks. Our research goal is to answer the key scientific question” How non-coding genetic variant act through cellular context specific gene regulatory network to influence disease”.
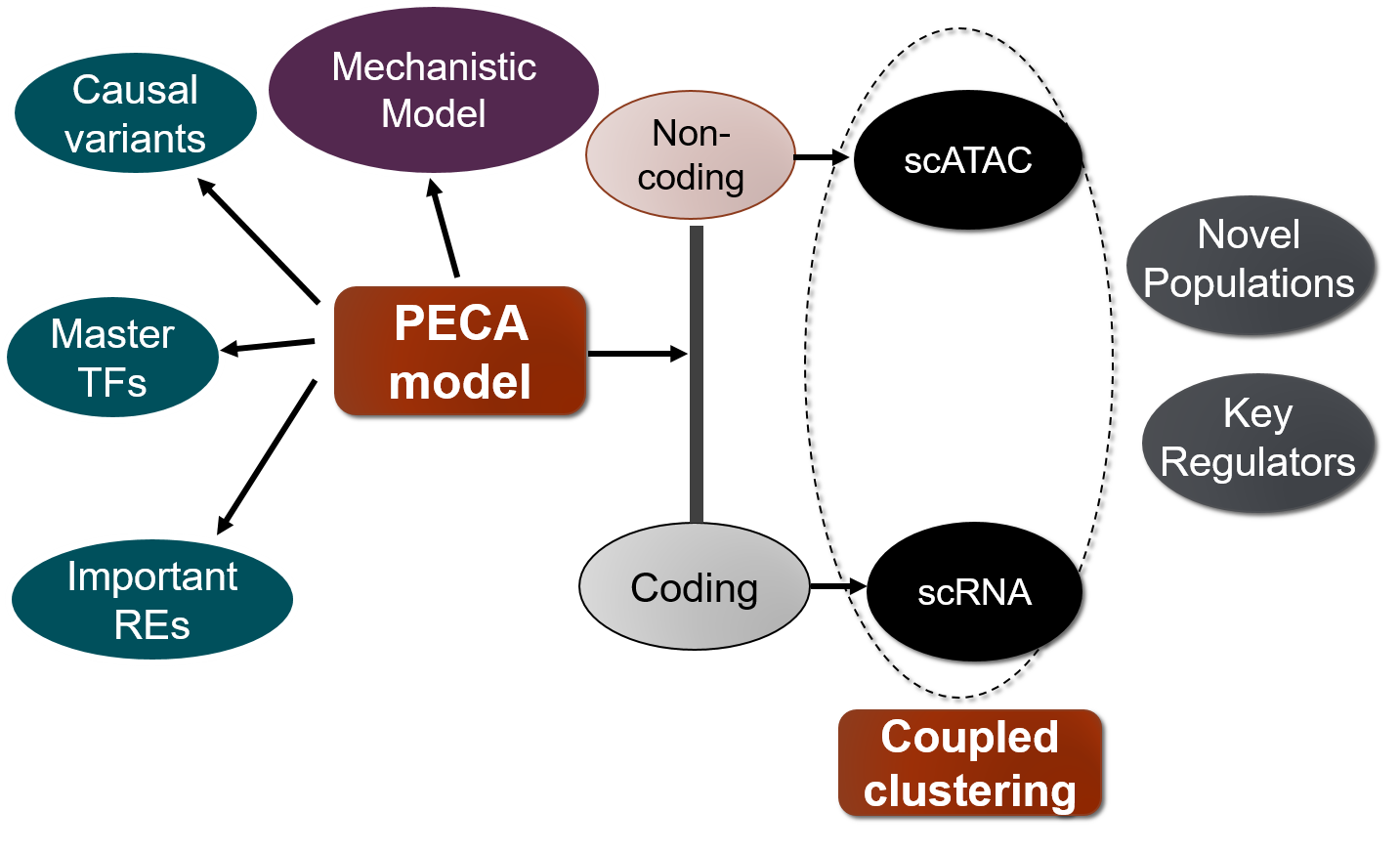
To do this, we are interested in developing novel statistical machine learning methods and bioinformatics tools including on following three directions.
1) Statistical modelling of gene regulatory network.
The inference of gene regulatory networks from experimental data is a central problem in computational biology. We are interested in constructing gene regulatory network by integrating different types of genomics data. We proposed a statistical inference model (Duren et al. 2017) for gene regulatory network inference from paired gene expression and chromatin accessibility data (PECA), which is the first mechanistic gene regulatory network model based on these two types of data integration. Further, we have extended this model to a time course experiment and developed a method named TimeReg (Duren et al. 2020). The transcriptional regulatory network inferred by PECA and TimeReg provides a detailed view of how trans- and cis-regulatory elements work together to affect gene expression in a context-specific manner. We applied this model on several collaboration projects and discovered promising results and validated by biological experiments. In the epidermal development stage, we discovered TFAP2C and P63 are the master regulator of lineage initiation and maturation (Li et al. 2019).
2) Integrative analysis of single-cell genomics data.
We are interested in methodogy of integrative analysis of single cell genomics data. We have proposed a model to perform coupled clustering (Duren et al. 2018) of scRNA-seq data and scATAC-seq data, which are from the same mixture context but different samples. We also extend this model to joint analysis of scRNA-seq, scATAC-seq, and bulk 3D chromatin contact data to perform deconvolution of bulk data and clustering of single cell data (Zeng et al 2019).
3) Annotation of non-coding genetic variants.
We are also interested in annotating genetic variants by cellular context specific gene regulatory networks. We proposed a method to prioritize noncoding variants based on their impact on context specific chromatin accessibility (Li et al. 2020). We also developed a variant interpretation methodology and apply it to the high-altitude adaption problem. In high-altitude adaptation, we discovered an important cis-regulatory element for EPAS1, and three SNPs on this element would affect the TF binding affinity and decrease the accessibility of regulatory element and target gene expression to make people better adapt to the high-altitude low-oxygen environment (Xin et al. 2020). We developed a Bayesian framework that integrates GWAS summary statistics with regulatory networks to infer genetic enrichments and associations simultaneously (Zhu et al. 2020).