Vision
We aim to make the regulome a computable system that explains how genomes shape phenotypes. Biomedicine is entering an inflection point where multimodal and spatial genomics make system-level regulatory modeling both feasible and necessary. We go beyond molecular readouts—markers, pathways, and differential signals—to build mechanistic, system-level models of regulation. Our lab develops element-resolved gene regulatory networks (GRNs) that link cis-regulatory elements, transcription factors, and target genes within cells, and place these networks in tissue context by modeling how cross-cell-type signaling modulates regulatory programs. By combining single-cell, multiomic, and spatial data, we identify driver regulatory mechanisms that underpin disease and generate testable hypotheses about where intervention could restore healthy programs.
All algorithms and methods developed in the lab can be accessed via our GitHub organization.
Research Pillars
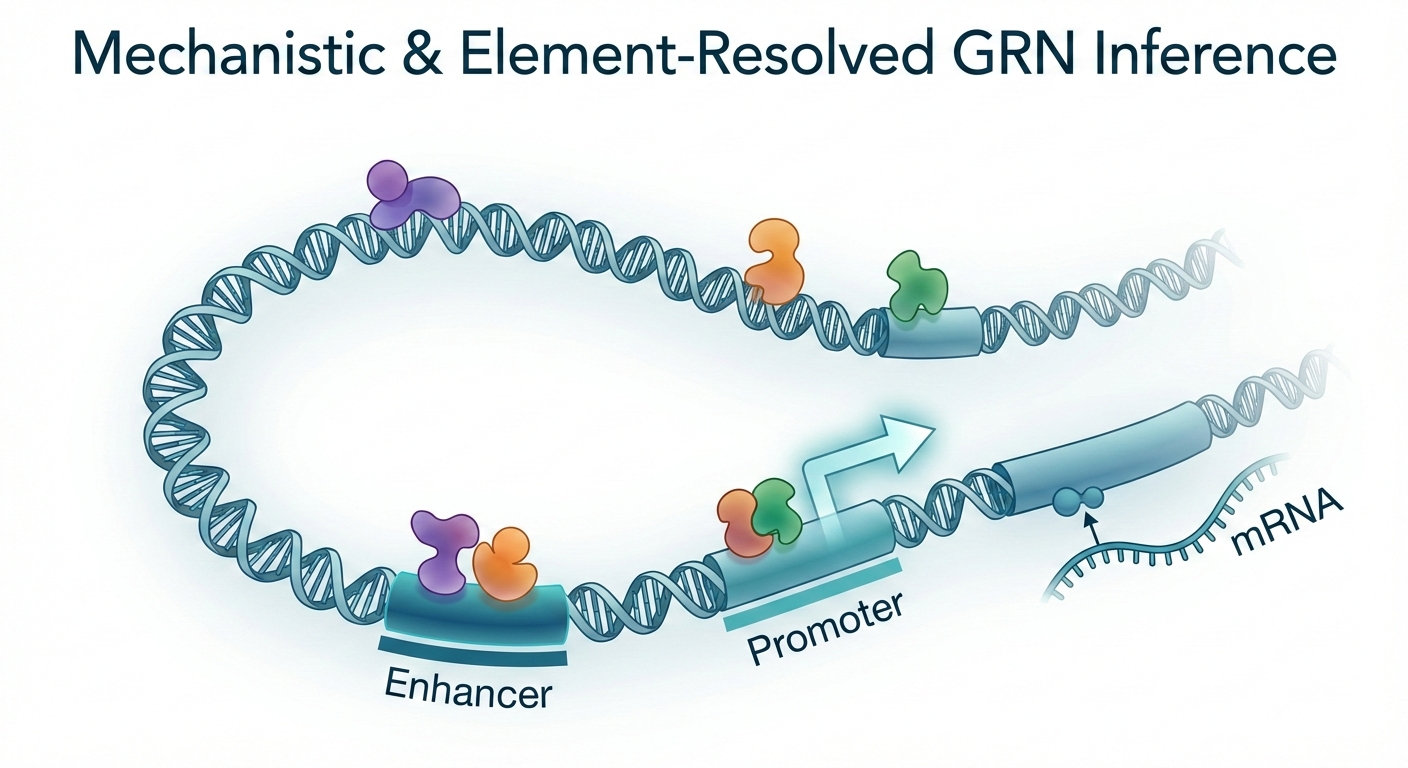
I. Mechanistic & Element-Resolved GRN Inference
We develop algorithms that model the physical and functional coupling between chromatin accessibility and gene expression, moving beyond simple correlations to resolve regulatory logic at the level of individual cis-elements and transcription factors. Our approach treats the genome not as a static map, but as a dynamic, coupled system.
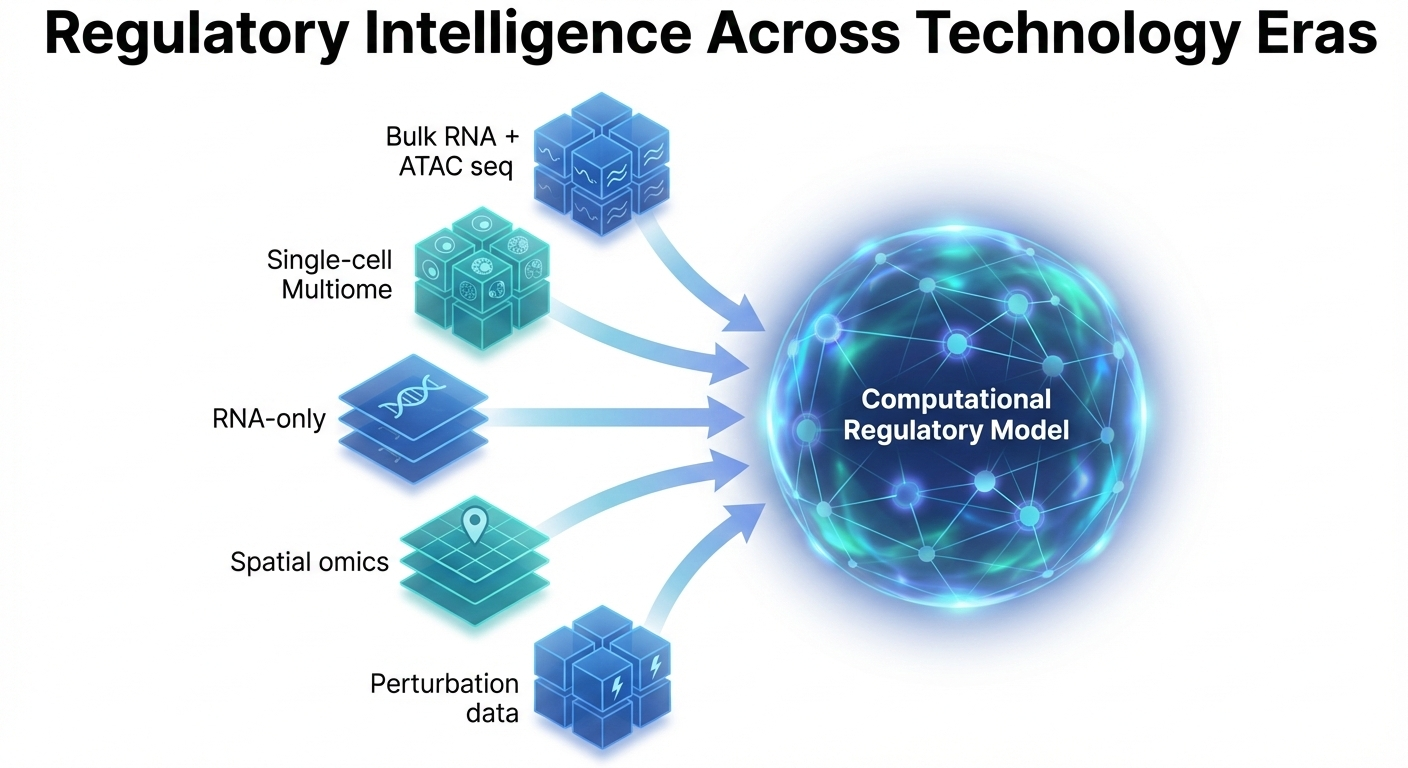
II. Regulatory Intelligence Across Technology Eras
We create “reusable” regulatory knowledge by developing frameworks that adapt pre-trained regulatory priors to new datasets. This bridges the gap between bulk, single-cell multiome, and RNA-only assays, enabling high-fidelity inference even when paired data is missing or noisy across changing experimental designs.
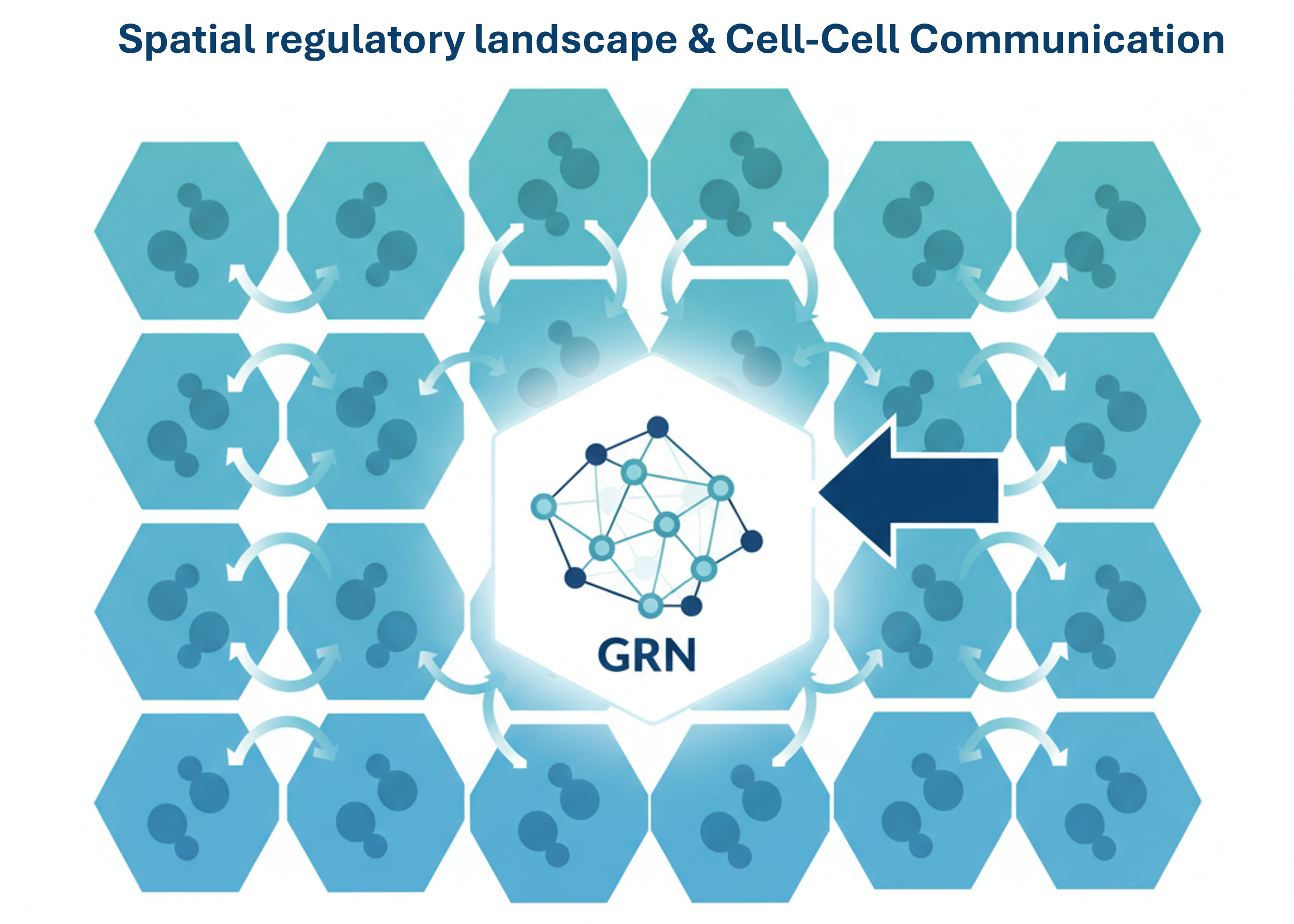
III. Spatial Regulatory Landscapes & Cell-Cell Communication
We place GRNs in their native tissue context to study how spatial organization and extracellular signaling (ligand-receptor interactions) rewire intracellular regulatory programs. By modeling cross-cell-type signaling, we understand how the tissue environment modulates gene expression.
IV. Regulatory Rewiring in Disease & Perturbations
We treat genetic variants, pharmacological treatments, and environmental stressors as “system-level rewiring events.” Our goal is to quantitatively predict how these factors shift regulatory programs and identify where intervention could restore healthy states.
